|
Scooped by
Gerd Moe-Behrens
onto SynBioFromLeukipposInstitute |

No comment yet.
Sign up to comment
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account


 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
From
www

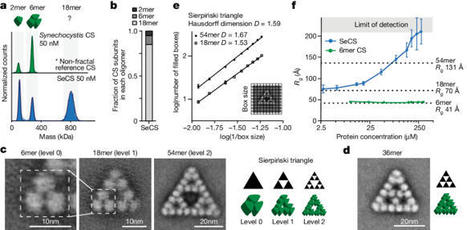
Fractals are patterns that are self-similar across multiple length-scales1. Macroscopic fractals are common in nature2–4; however, so far, molecular assembly into fractals is restricted to synthetic systems5–12. Here we report the discovery of a natural protein, citrate synthase from the cyanobacterium Synechococcus elongatus, which self-assembles into Sierpiński triangles. Using cryo-electron microscopy, we reveal how the fractal assembles from a hexameric building block. Although different stimuli modulate the formation of fractal complexes and these complexes can regulate the enzymatic activity of citrate synthase in vitro, the fractal may not serve a physiological function in vivo. We use ancestral sequence reconstruction to retrace how the citrate synthase fractal evolved from non-fractal precursors, and the results suggest it may have emerged as a harmless evolutionary accident. Our findings expand the space of possible protein complexes and demonstrate that intricate and regulatable assemblies can evolve in a single substitution. Citrate synthase from the cyanobacterium Synechococcus elongatus is shown to self-assemble into Sierpiński triangles, a finding that opens up the possibility that other naturally occurring molecular-scale fractals exist.
Prime editing enables the precise modification of genomes through reverse transcription of template sequences appended to the 3' ends of CRISPR-Cas guide RNAs1. To identify cellular determinants of prime editing, we developed scalable prime editing reporters and performed genome-scale CRI …
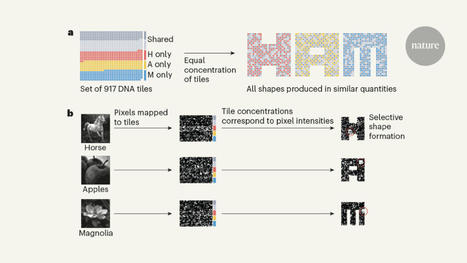
Self-assembling DNA can process information, but the computations have been limited to digital algorithms. A self-assembling DNA system has now been designed to perform complex pattern recognition.
Gerd Moe-Behrens's insight:
Here, we report the design, construction, and characterization of a tRNA neochromosome, a designer chromosome that functions as an additional, de novo counterpart to the native complement of Saccharomyces cerevisiae. Intending to address one of the central design principles of the Sc2.0 project, the ∼190-kb tRNA neochromosome houses all 275 relocated nuclear tRNA genes. To maximize stability, the design incorporates orthogonal genetic elements from non-S. cerevisiae yeast species. Furthermore, the presence of 283 rox recombination sites enables an orthogonal tRNA SCRaMbLE system. Following construction in yeast, we obtained evidence of a potent selective force, manifesting as a spontaneous doubling in cell ploidy. Furthermore, tRNA sequencing, transcriptomics, proteomics, nucleosome mapping, replication profiling, FISH, and Hi-C were undertaken to investigate questions of tRNA neochromosome behavior and function. Its construction demonstrates the remarkable tractability of the yeast model and opens up opportunities to directly test hypotheses surrounding these essential non-coding RNAs.
Gerd Moe-Behrens's insight:
The development of molecular couriers to selectively package, export, and recover RNA molecules within human cells is a significant challenge. In this issue of Cell, Horns et al.1 introduce cellular RNA exporters, termed COURIERs, that package, secrete, and protect RNA cargo and establish the foundation for sophisticated cell-to-cell RNA communication.
Gerd Moe-Behrens's insight:
Coevolution is a natural process by which two interacting proteins can change over time, resulting in different sequences at a conserved interface. Analysis of this process has been useful in protein structure prediction, and a deeper understanding of the mechanisms may help to augment the prediction of interfaces. Using a model protein complex, Yang et al. generated libraries of synthetic proteins with amino acids varying at six positions within a hydrophobic interface. A yeast display coevolution scheme allowed for the isolation of pairs of sequences that retained binding, resulting in a diverse collection of interactions based on the same starting scaffold. The authors then mapped the sequences in a coevolutionary network and determined structures of 10 pairs that provide details of specificity. They then used a pretrained protein language model to expand the scope of amino acid pairs, demonstrating the ability of this hybrid experimental-computational approach to give useful predictions for protein-protein interactions in this system. —MAF
Gerd Moe-Behrens's insight:
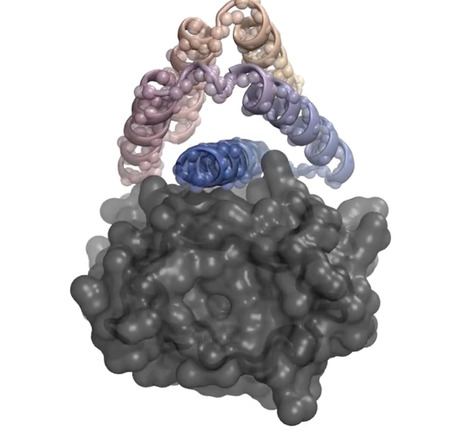
A new program could help scientists quickly design drugs and other helpful novel proteinsA team of U.S. researchers has created an artificial intelligence (AI) program capable of designing custom-tailored proteins that may speed efforts to design everything from drugs to fight cancer and infectious diseases to novel proteins able to quickly extract carbon dioxide from the atmosphere. The program, reported today in Nature, is called RFdiffusion. When asked to create possible drugs, it begins by analyzing a 3D model of a target protein, such as the insulin receptor seen here in gray, then designs novel proteins to bind to the target. The program starts off with random guesses as to the protein’s structure, then quickly homes in on tweaks to the architecture calculated to bind ever more tightly to the target. Compared with other non-AI protein design programs, the authors note, RFdiffusion improved the success rate of finding tight binders up to 100-fold.
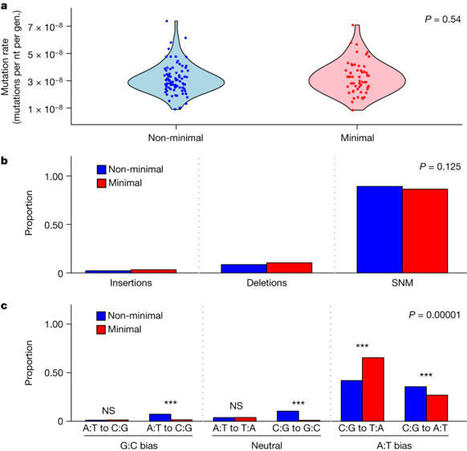
Possessing only essential genes, a minimal cell can reveal mechanisms and processes that are critical for the persistence and stability of life1,2. Here we report on how an engineered minimal cell3,4 contends with the forces of evolution compared with the Mycoplasma mycoides non-minimal cell from which it was synthetically derived. Mutation rates were the highest among all reported bacteria, but were not affected by genome minimization. Genome streamlining was costly, leading to a decrease in fitness of greater than 50%, but this deficit was regained during 2,000 generations of evolution. Despite selection acting on distinct genetic targets, increases in the maximum growth rate of the synthetic cells were comparable. Moreover, when performance was assessed by relative fitness, the minimal cell evolved 39% faster than the non-minimal cell. The only apparent constraint involved the evolution of cell size. The size of the non-minimal cell increased by 80%, whereas the minimal cell remained the same. This pattern reflected epistatic effects of mutations in ftsZ, which encodes a tubulin-homologue protein that regulates cell division and morphology5,6. Our findings demonstrate that natural selection can rapidly increase the fitness of one of the simplest autonomously growing organisms. Understanding how species with small genomes overcome evolutionary challenges provides critical insights into the persistence of host-associated endosymbionts, the stability of streamlined chassis for biotechnology and the targeted refinement of synthetically engineered cells2,7–9. An engineered minimal cell evolves to escape the negative consequences of genome streamlining.
Gerd Moe-Behrens's insight:
Terrestrial organisms developed circadian rhythms for adaptation to Earth’s quasi-24-h rotation. Achieving precise rhythms requires diurnal oscillation of fundamental biological processes, such as rhythmic shifts in the cellular translational landscape; however, regulatory mechanisms underlying rhythmic translation remain elusive. Here, we identified mammalian ATXN2 and ATXN2L as cooperating master regulators of rhythmic translation, through oscillating phase separation in the suprachiasmatic nucleus along circadian cycles. The spatiotemporal oscillating condensates facilitate sequential initiation of multiple cycling processes, from mRNA processing to protein translation, for selective genes including core clock genes. Depleting ATXN2 or 2L induces opposite alterations to the circadian period, whereas the absence of both disrupts translational activation cycles and weakens circadian rhythmicity in mice. Such cellular defect can be rescued by wild type, but not phase-separation-defective ATXN2. Together, we revealed that oscillating translation is regulated by spatiotemporal condensation of two master regulators to achieve precise circadian rhythm in mammals.
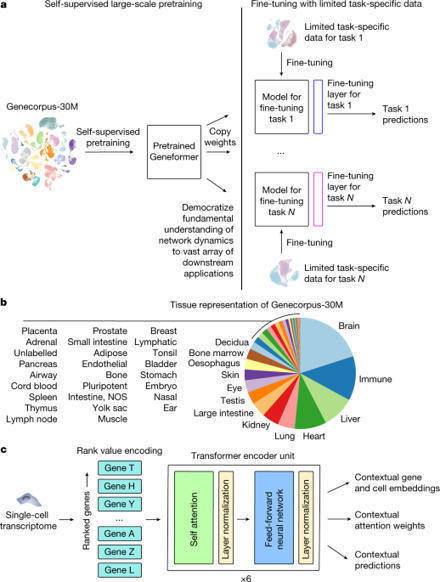
Mapping gene networks requires large amounts of transcriptomic data to learn the connections between genes, which impedes discoveries in settings with limited data, including rare diseases and diseases affecting clinically inaccessible tissues. Recently, transfer learning has revolutionized fields such as natural language understanding1,2 and computer vision3 by leveraging deep learning models pretrained on large-scale general datasets that can then be fine-tuned towards a vast array of downstream tasks with limited task-specific data. Here, we developed a context-aware, attention-based deep learning model, Geneformer, pretrained on a large-scale corpus of about 30 million single-cell transcriptomes to enable context-specific predictions in settings with limited data in network biology. During pretraining, Geneformer gained a fundamental understanding of network dynamics, encoding network hierarchy in the attention weights of the model in a completely self-supervised manner. Fine-tuning towards a diverse panel of downstream tasks relevant to chromatin and network dynamics using limited task-specific data demonstrated that Geneformer consistently boosted predictive accuracy. Applied to disease modelling with limited patient data, Geneformer identified candidate therapeutic targets for cardiomyopathy. Overall, Geneformer represents a pretrained deep learning model from which fine-tuning towards a broad range of downstream applications can be pursued to accelerate discovery of key network regulators and candidate therapeutic targets. A context-aware, attention-based deep learning model pretrained on single-cell transcriptomes enables predictions in settings with limited data in network biology and could accelerate discovery of key network regulators and candidate therapeutic targets.

Numerous real-world systems can be naturally modeled as multilayer networks, providing an efficient tool to characterize these complex systems. Although recent progress in understanding the controlling of synthetic multiplex networks, how to control real multilayer systems remains poorly understood. Here, we explore the controllability and energy requirement of molecular multiplex networks coupled by transcriptional regulatory network (TRN) and protein-protein interaction (PPI) network from the perspective of network structural characteristics. Our findings reveal that the driver nodes tend to avoid essential or pathogen-related genes. However, imposing external inputs on these essential or pathogen-related genes can remarkably reduce the energy cost, implying their crucial role in network control. Moreover, we find that the minimal driver nodes, as well as the energy required, are associated with disassortative coupling between TRN and PPI networks. Our results provide a comprehensive understanding of the roles of genes in biology and network control across several species.
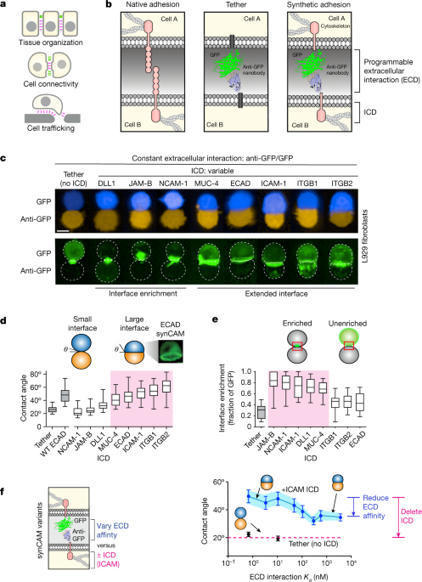
Cell adhesion molecules are ubiquitous in multicellular organisms, specifying precise cell–cell interactions in processes as diverse as tissue development, immune cell trafficking and the wiring of the nervous system1–4. Here we show that a wide array of synthetic cell adhesion molecules can be generated by combining orthogonal extracellular interactions with intracellular domains from native adhesion molecules, such as cadherins and integrins. The resulting molecules yield customized cell–cell interactions with adhesion properties that are similar to native interactions. The identity of the intracellular domain of the synthetic cell adhesion molecules specifies interface morphology and mechanics, whereas diverse homotypic or heterotypic extracellular interaction domains independently specify the connectivity between cells. This toolkit of orthogonal adhesion molecules enables the rationally programmed assembly of multicellular architectures, as well as systematic remodelling of native tissues. The modularity of synthetic cell adhesion molecules provides fundamental insights into how distinct classes of cell–cell interfaces may have evolved. Overall, these tools offer powerful abilities for cell and tissue engineering and for systematically studying multicellular organization. Synthetic cell adhesion molecules yield customized cell–cell interactions with adhesion properties that are similar to native interactions, and offer abilities for cell and tissue engineering and for systematically studying multicellular organization.
Gerd Moe-Behrens's insight:
Parsing nodulation pathwaysLegumes benefit from symbiotic microbes resident in root nodules that fix nitrogen, whereas most other plants depend on externally supplied or gathered nitrogen. Rübsam et al. identified a bifunctional receptor complex in the model legume Lotus japonicus that initiates development and infection of the root nodules. Development of root nodules could be driven solely by the intracellular domains containing the kinases once the receptor complex was formed, but the Nod factor–recognizing ectodomains were required to support rhizobial infection. Receptors identified in the cereal barley, which does not form symbiotic nitrogen-fixing root nodules, were able to support Lotus root nodule organogenesis, suggesting that the path to nitrogen fixation in cereals might be shorter than we thought. —PJH AbstractUnderstanding the composition and activation of multicomponent receptor complexes is a challenge in biology. To address this, we developed a synthetic approach based on nanobodies to drive assembly and activation of cell surface receptors and apply the concept by manipulating receptors that govern plant symbiosis with nitrogen-fixing bacteria. We show that the Lotus japonicus Nod factor receptors NFR1 and NFR5 constitute the core receptor complex initiating the cortical root nodule organogenesis program as well as the epidermal program controlling infection. We find that organogenesis signaling is mediated by the intracellular kinase domains whereas infection requires functional ectodomains. Finally, we identify evolutionarily distant barley receptors that activate root nodule organogenesis, which could enable engineering of biological nitrogen-fixation into cereals.
|

From
www
" Treatment with chimeric antigen receptor (CAR) T cells targeting the B-cell maturation antigen (BCMA) has shown remarkable results in patients with relapsed or refractory multiple myeloma (MM). However, most patients eventually relapse, underscoring the need for improved therapeutic approaches. Now, Díez-Alonso et al. have engineered T cells to secrete T cell–engaging (TCE) antibodies targeting BCMA on cancer cells and CD3 on T cells. In mouse models of MM, these TCE antibody secreting (STAb) T cells were more effective at eliminating target cells than traditional CAR-T cells......"

From
www
Artificial electron donors and acceptors expand researchers’ metabolic engineering options — if only cells would cooperate.
The Construction File (CF) specification establishes a standardized interface for molecular biology operations, laying a foundation for automation and enhanced efficiency in experiment design. It is implemented across three distinct software projects: PyDNA_CF_Simulator, a Python project featuring a …
|
|
|
Scooped by Gerd Moe-Behrens |
Recent advances in structural DNA nanotechnology have been facilitated by design tools that continue to push the limits of structural complexity while simplifying an often-tedious design process. We recently introduced the software MagicDNA, which enables design of complex 3D DNA assemblies with many components; however, the design of structures with free-form features like vertices or curvature still required iterative design guided by simulation feedback and user intuition. Here, we present an updated design tool, MagicDNA 2.0, that automates the design of free-form 3D geometries, leveraging design models informed by coarse-grained molecular dynamics simulations. Our GUI-based, stepwise design approach integrates a high level of automation with versatile control over assembly and subcomponent design parameters. We experimentally validated this approach by fabricating a range of DNA origami assemblies with complex free-form geometries, including a 3D Nozzle, G-clef, and Hilbert and Trifolium curves, confirming excellent agreement between design input, simulation, and structure formation.
|
|
Scooped by Gerd Moe-Behrens |
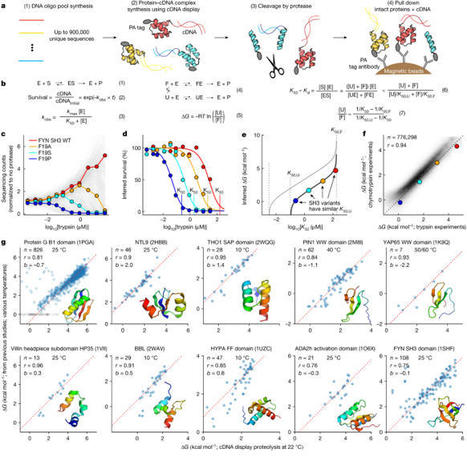
Advances in DNA sequencing and machine learning are providing insights into protein sequences and structures on an enormous scale1. However, the energetics driving folding are invisible in these structures and remain largely unknown2. The hidden thermodynamics of folding can drive disease3,4, shape protein evolution5–7 and guide protein engineering8–10, and new approaches are needed to reveal these thermodynamics for every sequence and structure. Here we present cDNA display proteolysis, a method for measuring thermodynamic folding stability for up to 900,000 protein domains in a one-week experiment. From 1.8 million measurements in total, we curated a set of around 776,000 high-quality folding stabilities covering all single amino acid variants and selected double mutants of 331 natural and 148 de novo designed protein domains 40–72 amino acids in length. Using this extensive dataset, we quantified (1) environmental factors influencing amino acid fitness, (2) thermodynamic couplings (including unexpected interactions) between protein sites, and (3) the global divergence between evolutionary amino acid usage and protein folding stability. We also examined how our approach could identify stability determinants in designed proteins and evaluate design methods. The cDNA display proteolysis method is fast, accurate and uniquely scalable, and promises to reveal the quantitative rules for how amino acid sequences encode folding stability. Large-scale assays using cDNA display proteolysis are used to measure the folding stabilities of protein domains, providing a method to quantify the effects of mutations on protein folding, with applications in protein design.
|
|
Scooped by Gerd Moe-Behrens |
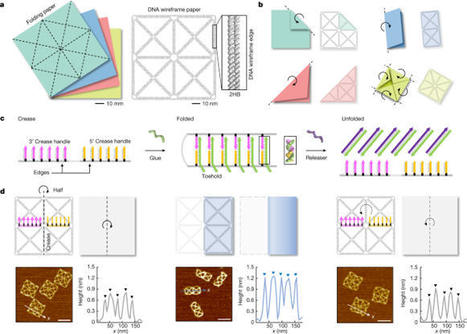
The paper-folding mechanism has been widely adopted in building of reconfigurable macroscale systems because of its unique capabilities and advantages in programming variable shapes and stiffness into a structure1–5. However, it has barely been exploited in the construction of molecular-level systems owing to the lack of a suitable design principle, even though various dynamic structures based on DNA self-assembly6–9 have been developed10–23. Here we propose a method to harness the paper-folding mechanism to create reconfigurable DNA origami structures. The main idea is to build a reference, planar wireframe structure24 whose edges follow a crease pattern in paper folding so that it can be folded into various target shapes. We realized several paper-like folding and unfolding patterns using DNA strand displacement25 with high yield. Orthogonal folding, repeatable folding and unfolding, folding-based microRNA detection and fluorescence signal control were demonstrated. Stimuli-responsive folding and unfolding triggered by pH or light-source change were also possible. Moreover, by employing hierarchical assembly26 we could expand the design space and complexity of the paper-folding mechanism in a highly programmable manner. Because of its high programmability and scalability, we expect that the proposed paper-folding-based reconfiguration method will advance the development of complex molecular systems. A method is presented to harness the paper-folding mechanism of reconfigurable macroscale systems to create reconfigurable DNA origami structures, in anticipation that it will advance the development of complex molecular systems.
|
|
Scooped by Gerd Moe-Behrens |
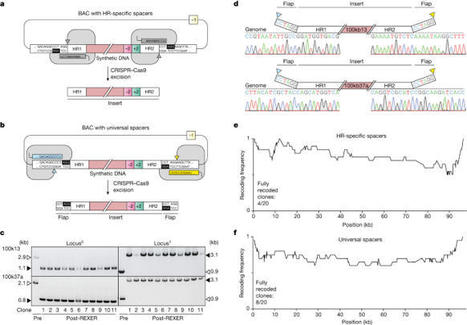
Whole-genome synthesis provides a powerful approach for understanding and expanding organism function1–3. To build large genomes rapidly, scalably and in parallel, we need (1) methods for assembling megabases of DNA from shorter precursors and (2) strategies for rapidly and scalably replacing the genomic DNA of organisms with synthetic DNA. Here we develop bacterial artificial chromosome (BAC) stepwise insertion synthesis (BASIS)—a method for megabase-scale assembly of DNA in Escherichia coli episomes. We used BASIS to assemble 1.1 Mb of human DNA containing numerous exons, introns, repetitive sequences, G-quadruplexes, and long and short interspersed nuclear elements (LINEs and SINEs). BASIS provides a powerful platform for building synthetic genomes for diverse organisms. We also developed continuous genome synthesis (CGS)—a method for continuously replacing sequential 100 kb stretches of the E. coli genome with synthetic DNA; CGS minimizes crossovers1,4 between the synthetic DNA and the genome such that the output for each 100 kb replacement provides, without sequencing, the input for the next 100 kb replacement. Using CGS, we synthesized a 0.5 Mb section of the E. coli genome—a key intermediate in its total synthesis1—from five episomes in 10 days. By parallelizing CGS and combining it with rapid oligonucleotide synthesis and episome assembly5,6, along with rapid methods for compiling a single genome from strains bearing distinct synthetic genome sections1,7,8, we anticipate that it will be possible to synthesize entire E. coli genomes from functional designs in less than 2 months. BAC stepwise insertion synthesis (BASIS) can be used to build synthetic genomes for diverse organisms, and continuous genome synthesis (CGS) enables the rapid synthesis of entire Escherichia coli genomes from functional designs.
|
|
Scooped by Gerd Moe-Behrens |
By modifying the blueprint of life, researchers are endowing proteins with chemistries they’ve never had before.
|
|
Scooped by Gerd Moe-Behrens |
Pretraining on a large data set gives model insight into how genes interact.
A deep-learning model called Geneformer has been developed and pretrained using about 30 million single-cell gene-expression profiles to enable it to make predictions about gene-network biology in instances in which gene-expression data are limited. Geneformer can be tuned for many downstream applications to accelerate discovery of key gene-network regulators and candidate therapeutic targets.
|
|
Scooped by Gerd Moe-Behrens |
Enzyme function annotation is a fundamental challenge, and numerous computational tools have been developed. However, most of these tools cannot accurately predict functional annotations, such as enzyme commission (EC) number, for less-studied proteins or those with previously uncharacterized functions or multiple activities. We present a machine learning algorithm named CLEAN (contrastive learning–enabled enzyme annotation) to assign EC numbers to enzymes with better accuracy, reliability, and sensitivity compared with the state-of-the-art tool BLASTp. The contrastive learning framework empowers CLEAN to confidently (i) annotate understudied enzymes, (ii) correct mislabeled enzymes, and (iii) identify promiscuous enzymes with two or more EC numbers—functions that we demonstrate by systematic in silico and in vitro experiments. We anticipate that this tool will be widely used for predicting the functions of uncharacterized enzymes, thereby advancing many fields, such as genomics, synthetic biology, and biocatalysis.
|
|
Scooped by Gerd Moe-Behrens |
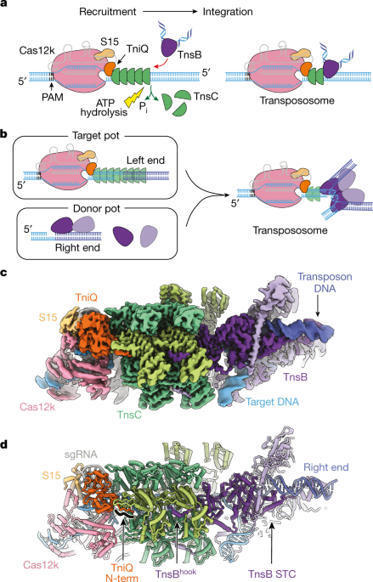
CRISPR-associated transposons (CAST) are programmable mobile genetic elements that insert large DNA cargos using an RNA-guided mechanism1–3. CAST elements contain multiple conserved proteins: a CRISPR effector (Cas12k or Cascade), a AAA+ regulator (TnsC), a transposase (TnsA–TnsB) and a target-site-associated factor (TniQ). These components are thought to cooperatively integrate DNA via formation of a multisubunit transposition integration complex (transpososome). Here we reconstituted the approximately 1 MDa type V-K CAST transpososome from Scytonema hofmannii (ShCAST) and determined its structure using single-particle cryo-electon microscopy. The architecture of this transpososome reveals modular association between the components. Cas12k forms a complex with ribosomal subunit S15 and TniQ, stabilizing formation of a full R-loop. TnsC has dedicated interaction interfaces with TniQ and TnsB. Of note, we observe TnsC–TnsB interactions at the C-terminal face of TnsC, which contribute to the stimulation of ATPase activity. Although the TnsC oligomeric assembly deviates slightly from the helical configuration found in isolation, the TnsC-bound target DNA conformation differs markedly in the transpososome. As a consequence, TnsC makes new protein–DNA interactions throughout the transpososome that are important for transposition activity. Finally, we identify two distinct transpososome populations that differ in their DNA contacts near TniQ. This suggests that associations with the CRISPR effector can be flexible. This ShCAST transpososome structure enhances our understanding of CAST transposition systems and suggests ways to improve CAST transposition for precision genome-editing applications. Structural studies of the CRISPR-associated transposon comprising Cas12k, TnsC, TnsB and TniQ from Scytonema hofmannii using cryo-electron microscopy reveal insights into the architecture and mechanism of RNA-guided DNA transposition.